-
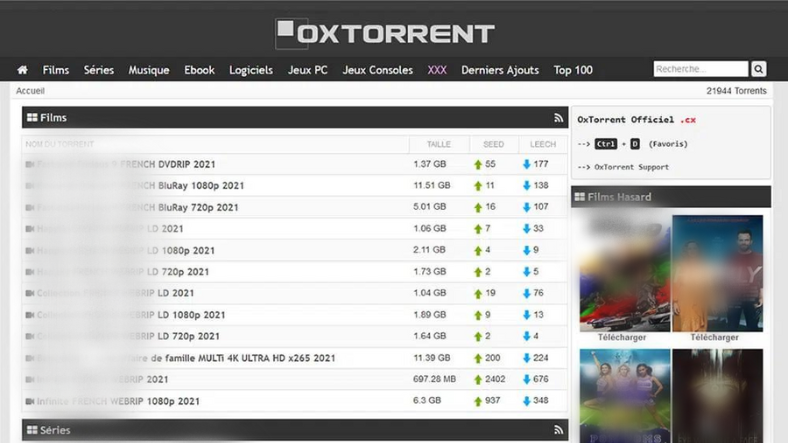
Changer l’adresse du site OxTorrent : comment y accéder désormais ?
Comment accéder à OxTorrent malgré les changements d’adresse? OxTorrent est un site populaire de téléchargement de torrents qui offre un large choix de fichiers à ses utilisateurs. Au fil du temps, ce site a connu plusieurs changements d’adresse, ce qui peut causer des problèmes d’accès pour les utilisateurs réguliers. Si vous êtes un adepte d’OxTorrent et que vous cherchez des solutions pour accéder au site malgré les changements d’adresse, voici quelques astuces qui pourraient vous aider. Partie 1: Les différentes adresses pour accéder à OxTorrent S’il est vrai qu’OxTorrent a subi plusieurs changements d’adresse, il existes plusieurs adresses actuelles pour y accéder. Selon les utilisateurs, les adresses suivantes sont les…
-
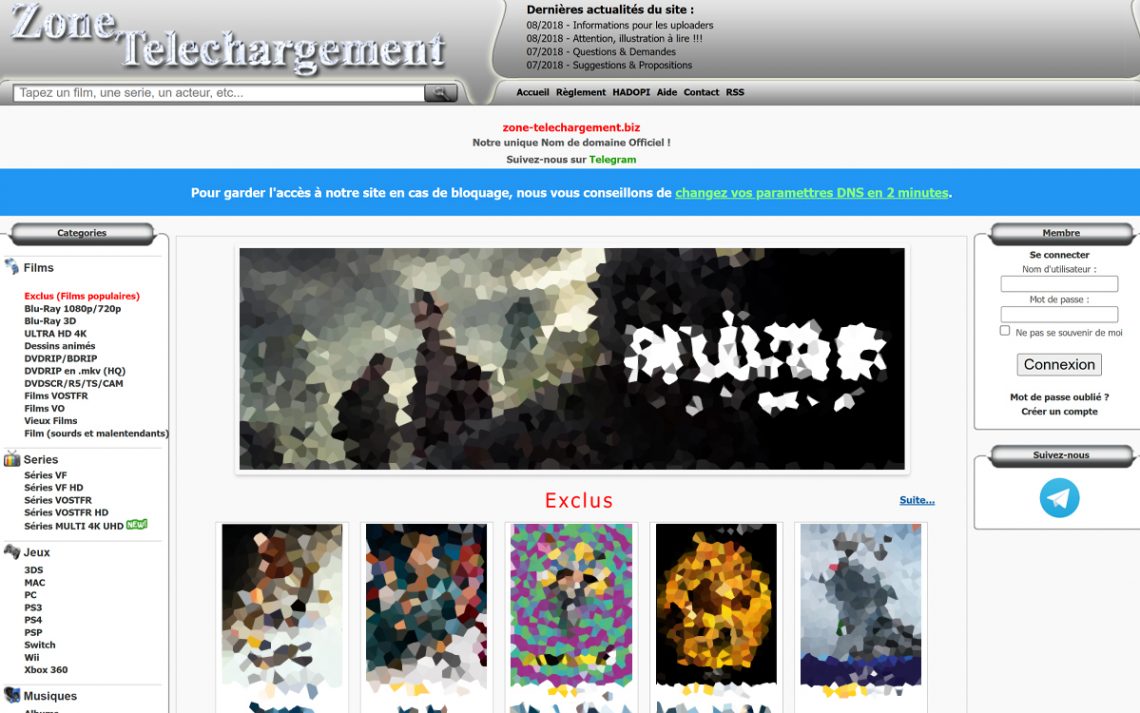
Nouvelle adresse officielle de Zone-Téléchargement : où la trouver ?
Zone-Téléchargement : bref historique, fermeture, retour et nouvelle adresse officielle Introduction Depuis sa création en 2012, Zone-Téléchargement est devenu un site de téléchargement de contenus numériques très populaire en France. Les éléments accessibles comprenaient les films, les séries, les jeux, la musique, les logiciels et bien plus encore, et tout cela gratuitement et sans limite de téléchargement. Toutefois, en novembre 2016, le site a été contraint de fermer après une décision de justice. Les utilisateurs ont donc été contraints de s’adapter à une nouvelle adresse URL s’ils souhaitaient continuer à accéder aux éléments proposés.
-
Comprendre le fonctionnement de WawaCity et pourquoi son adresse URL a changé.
Introduction Bienvenue sur mon article consacré au site WawaCity. Dans cet article, je vais vous présenter l’historique, le fonctionnement et les raisons qui ont poussé WawaCity à changer d’adresse URL. Historique de WawaCity WawaCity est un site de téléchargement direct créé en 2009. Au fil des années, le site a connu une croissance rapide et a évolué pour devenir l’un des sites de référence pour les téléchargements directs. Cependant, cette croissance rapide a également apporté son lot de difficultés et de problèmes. En effet, le site a dû faire face à un grand nombre de problèmes de droits d’auteur et a souvent été accusé de mettre à disposition des contenus…
-
Les 30 accessoires de cuisine les plus innovants
-
Top des aspirateurs balais laveurs polyvalents 2-en-1 et 3-en-1
You May Also Like

Essayons de reformuler le titre de l’article pour une autre plateforme : Vorwerk lance le Kobold VB 100, un aspirateur-laveur sans fil ultra-léger de qualité supérieure.

Comparaison entre l’aspirateur laveur Tineco Floor One S5 Combo et ses concurrents : revue et essai complet.

Les meilleurs aspirateurs-balais comparés : notre sélection
-
Meilleures options de trottinettes électriques pour vous déplacer après la fin du service en libre-service – Top 16 de la liste.
Guide d’achat 2021 des meilleures trottinettes électriques L’utilisation précédente des trottinettes électriques en libre-service a connu une fin abrupte, et, aujourd’hui, de plus en plus d’utilisateurs préfèrent se procurer leur propre équipement. Avec l’augmentation de la demande, le marché des trottinettes électriques s’est considérablement développé et offre aujourd’hui une multitude d’options pour tous les besoins et budgets. Nous avons compilé une liste des seize meilleures trottinettes électriques du marché afin que vous puissiez trouver celle qui correspond le mieux à vos besoins personnels.
You May Also Like

Unagi, la marque qui révolutionne les trottinettes électriques, présente son dernier modèle au design futuriste.

Les boutiques les plus recommandées pour acheter une trottinette électrique.

Comparaison des trottinettes électriques Wegoboard Suprem 3, Xiaomi Mi 3 et Xiaomi Mi Essential pour vous aider à choisir la meilleure option.
-
Découvrez MYM, la plateforme de vidéos exclusives de célébrités qui fait sensation !
-
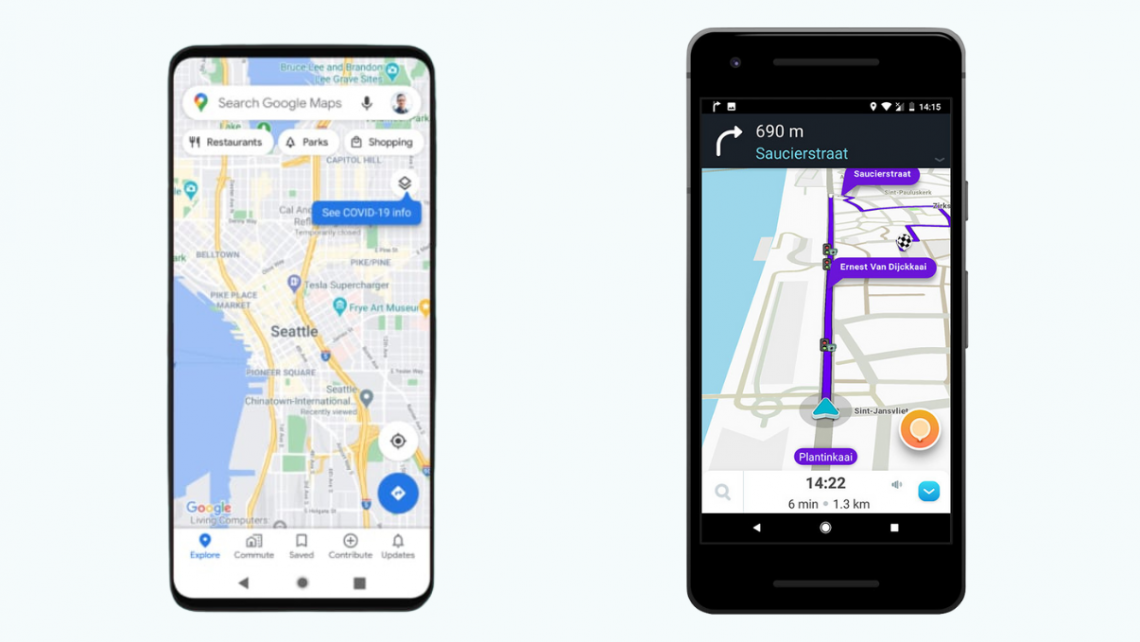
Comparaison entre Waze et Google Maps : quel est le meilleur GPS gratuit pour téléphone portable ?
Comparaison entre Waze et Google Maps: Choisir le meilleur GPS pour votre Smartphone Lorsqu’il s’agit de naviguer dans une ville, un bon GPS sur votre smartphone est essentiel. Avec autant d’options sur le marché, choisir la meilleure application pour vos besoins peut être un défi. Dans cet article, nous allons comparer deux des applications de navigation les plus populaires: Waze et Google Maps, afin de vous aider à prendre la meilleure décision pour votre prochain voyage. Interface Utilisateur Waze et Google Maps ont tous deux des interfaces utilisateur distinctes. Waze, avec ses icônes ludiques et sa fonctionnalité d’affichage de trafic en temps réel, a une apparence conviviale et facile à…
-
Où trouver la nouvelle adresse officielle de Wiflix ?
-
Les lunettes connectées et AR les plus performantes à découvrir sur le marché et à venir.
Introduction Les lunettes connectées et AR, une technologie en pleine expansion, réunissant connectivité et réalité augmentée. Dans cet article, nous allons passer en revue les différentes lunettes connectées et AR sur le marché, les caractéristiques de chaque produit ainsi que les perspectives futures de cette technologie.